Comment on:
Widespread purifying selection on RNA structure in mammals
Martin Smith, Tanja Gesell, Peter F. Stadler, and John Mattick. Nucleic Acid
Research. 2013.
Source |
Archive.is
In 2013, John Mattick and three other biologists published a study comparing RNA structure across mammals, finding that 13.6% to 30% of it was conserved independent of DNA sequence:
When applied to consistency-based multiple genome alignments of 35 [placental and marsupial, including including bats, mice, pigs, cows, dolphins and human] mammals, our approach confidently identifies >4 million evolutionarily constrained RNA structures using a conservative sensitivity threshold that entails historically low false discovery rates for such analyses (5–22%). These predictions comprise 13.6% of the human genome, 88% of which fall outside any known sequence-constrained element, suggesting that a large proportion of the mammalian genome is functional.
...
Our findings provide an additional layer of support for previous reports advancing that >20% of the human genome is subjected to evolutionary selection while suggesting that additional evidence for function can be uncovered through careful investigation of analytically involute higher-order RNA structures. Furthermore, our approach entails an empirically determined false discovery rate well below that reported in previous endeavors (i.e. 5–22% versus 50–70%), supporting the widespread involvement of RNA secondary structure in mammalian evolution.
...
the RNA structure predictions we report using conservative thresholds are likely to span >13.6% of the human genome we report. This number is probably a substantial underestimate of the true proportion given the conservative scoring thresholds employed, the neglect of pseudoknots, the liberal distance between overlapping windows and the incapacity of the sliding-window approach to detect base-pair interactions outside the fixed window length. A less conservative estimate would place this ratio somewhere above 20% from the reported sensitivities measured from native RFAM alignments and over 30% from the observed sensitivities derived from sequence-based realignment of RFAM data. Our data complement recent findings from the ENCODE consortium, which report that 74.7% of the human genome is transcribed in multiple cell lines and that many novel unannotated genes are detected when sequencing RNA from subcellular compartments.
...
The resulting ECS [evolutionarily conserved structure] predictions encompass 18.5% of the surveyed alignments that, in turn, span across 84.1% of the human genome.
...
With regards to the genomic distribution of hits, the majority of predictions lie within intronic and intergenic regions [see figure 4B]... Predictions are roughly 2-fold enriched (odds ratio) in annotated exons, with the highest enrichment observed in protein-coding regions... about half of the ECS predictions we report overlap repeat elements... The prevalence of ECS structures is strongly enriched in Alu elements, which are known to form conserved RNA secondary structures.
...
the majority (87.8%) of the ECS predictions reported herein lie outside annotated sequence-constrained elements
The paper was long and highly technical, but thankfully it was summarized in a press release:
While other studies have shown that around 5-8% of the genome is conserved at the level of DNA sequence, indicating that it is functional, the new study shows that in addition much more, possibly up to 30%, is also conserved at the level of RNA structure.
Conserved sequences (those shared with other mammals) only provide a lower estimate to how much is functional. They also comment that RNA's require a specific structure to be functional:
the nucleic acids that make up RNA connect to each other in very specific ways, which force RNA molecules to twist and loop into a variety of complicated 3D structures.
But how could RNA secondary structure be conserved if the DNA encoding for it is not? Analysis of the fitness effect of compensatory mutations (HSFP J, 2009) may explain:
It is well known that the folding of RNA molecules into the stem-loop structure requires base pair matching in the stem part of the molecule, and mutations occurring to one segment of the stem part will disrupt the matching, and therefore, have a deleterious effect on the folding and stability of the molecule. It has been observed that mutations in the complementary segment can rescind the deleterious effect by mutating into a base pair that matches the already mutated base, thus recovering the fitness of the original molecule (Kelley et al., 2000; Wilke et al., 2003).
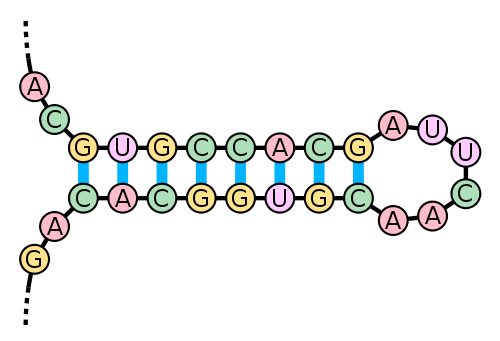
This diagram from Wikipedia (CC BY-SA 3.0 license) helps illustrate. If a base on the top mutates, a base on the bottom can also mutate to match it again, which maintains the same secondary structure.